Una ricerca del team dell’Università di Trieste pubblicata sulla rivista scientifica Scientific Reports ha studiato gli effetti delle varianti circolanti di SARS-CoV-2 sull’interazione con degli anticorpi monoclonali utilizzati in terapia. Attraverso l’uso di tecniche di simulazione molecolare sono stati identificati i residui aminoacidici del virus che, una volta mutati, possono causare una drastica diminuzione dell’efficacia terapeutica di questi agenti.
Possibili applicazioni nella previsione dell’efficacia e nello sviluppo di nuove terapie antivirali.
Uno studio di un gruppo di ricercatori dell’Università di Trieste pubblicato sulla rivista Scientific Reports (Springer Nature) ha utilizzato un approccio computazionale per prevedere gli effetti delle varianti di SARS-CoV-2 sull’efficacia terapeutica di due anticorpi monoclonali, bamlanivimab e etesevimab. Gli anticorpi monoclonali sono derivati, tramite particolari procedure di laboratorio, da molecole che il nostro organismo produce naturalmente in risposta ad un’infezione o dopo la somministrazione di un vaccino. I due anticorpi somministrati insieme sono autorizzati dallo scorso febbraio per il trattamento di COVID-19 da lieve a moderato, sia negli Stati Uniti che in Europa.
In particolare, la metodologia applicata in questo studio ha permesso di prevedere e spiegare a livello molecolare gli effetti negativi sull’attività di questi agenti terapeutici di sostituzioni nelle posizioni 452 e 484 di SARS-Cov-2, mutazioni presenti nella ben nota variante delta (indiana).
In generale, i risultati computazionali presentati, fornendo un razionale molecolare agli effetti delle varianti circolanti di SARS-CoV-2, costituiscono uno strumento rapido e affidabile per identificare i punti di forza e di debolezza delle terapie anticorpali nei confronti di un virus in continua evoluzione fornendo quindi sostanziali informazioni strutturali per lo sviluppo di anticorpi più efficienti.
Sono questi i risultati ottenuti dal team di ricerca Molecular Biology and Nanotechnology Laboratory (MolBNL@UniTS) operativo al Dipartimento di Ingegneria e Architettura dell’Università degli studi di Trieste, che ha ottenuto immediato interesse tra gli scienziati che stanno combattendo contro COVID-19.
“Rispetto ai precedenti studi eseguiti dal nostro gruppo di ricerca sulle varianti di SARS-CoV-2 – spiega Erik Laurini, co-fondatore del gruppo MolBNL@UniTS – in questo lavoro abbiamo spostato il punto di vista. Prima guardavamo l’effetto di mutanti sull’interazione con la proteina umana che il virus sfrutta per entrare nelle nostre cellule. In quel caso andavamo a prevedere un eventuale aumento della pericolosità e dell’infettività del virus. In questo articolo, invece, siamo andati a valutare il ruolo della diversità genetica virale sull’efficacia di uno degli attuali trattamenti terapeutici. Prevedere in tempi rapidi se una nuova mutazione può compromettere l’efficacia di un agente antivirale sarà fondamentale per combattere definitivamente il COVID-19”.
Per condurre la ricerca sono state adottate delle tecniche di simulazione al calcolatore nell’ambito dell’High Performance Computing (HPC) che hanno permesso l’impiego di risorse e investimenti ridotti, a fronte di un’altissima rapidità di processazione dei dati con tempi non paragonabili a quelli dei laboratori sperimentali.
“Il prossimo obiettivo sarà quello di sfruttare le informazioni raccolte per costruire un modello computazionale in grado di progettare anticorpi monoclonali resistenti alle varianti più pericolose di SARS-Cov-2 - dichiara Domenico Marson, assegnista del gruppo MolBNL@UniTS - Questi farmaci innovativi, infatti, possono essere modificati nella loro struttura in maniera tale da sopperire all’interferenza provocata da una mutazione virale mantenendo allo stesso tempo le altre interazioni che ne garantivano l’efficacia”.
Lo studio può avere importanti applicazioni nella previsione dell’efficacia di vaccini e terapie come altri tipi di anticorpi monoclonali e farmaci antivirali. L’utilizzo del sistema studiato dal team triestino consentirà di valutare questi e altri rimedi in maniera più veloce ed efficace.
“È importante sottolineare che la procedura computazionale descritta in questo articolo ha un carattere veramente generale. - spiega Sabrina Pricl, coordinatrice del gruppo MolBNL@UnitTS– Infatti, può essere applicata per prevedere in modo rapido ed affidabile l'effetto delle mutazioni anche su altri sistemi di interazione proteina/proteina, così come proteina/ligando e proteina/acido nucleico, che giocano ruoli chiave nella patogenesi di importanti malattie umane tra cui, ad esempio, infezioni batteriche, sindromi ereditarie e, soprattutto il cancro, come già dimostrato in precedenti studi dal nostro gruppo di ricerca. L'HPC, inoltre, è una risorsa strategica per il futuro dell'Europa nel campo delle nanobiotecnologie. Costituisce, infatti, un pilastro dei programmi di finanziamento Europei sia nei precedenti Horizon 2020 che in quelli attuali Horizon Europe”.
Studio pubblicato su su Scientific Reports (Springer Nature), 12 Ottobre 2021
Molecular rationale for SARS‑CoV‑2 spike circulating mutations able to escape bamlanivimab and etesevimab monoclonal antibodies
Erik Laurini, Domenico Marson, Suzana Aulic, Alice Fermeglia, and Sabrina Pricl
Molecular Biology and Nanotechnology Laboratory (MolBNL@UniTS), DEA,
Università degli studi di Trieste
https://doi.org/10.1038/s41598-021-99827-3
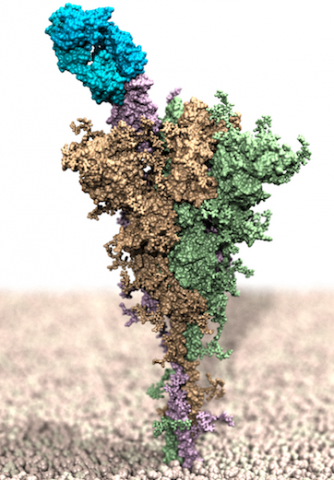
Nell'immagine a fianco: Rendering al computer della proteina Spike di SARS-CoV-2 a lunghezza intera, incorporato in un modello di membrana ed in complesso con l'anticorpo monoclonale bamlanivimab (in azzurro).